
Foto: Mihai Mănăilă
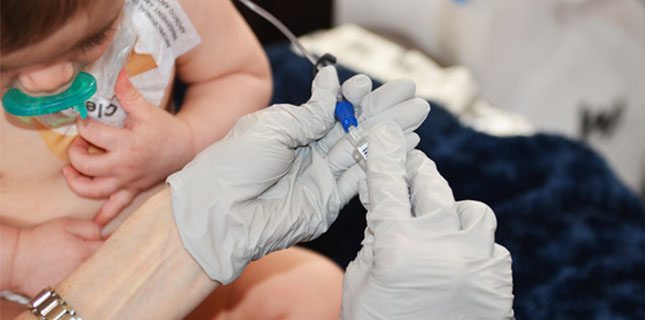
Medicul citeşte noul set de analize. Ridică neputincios din umeri. „Da, transaminazele sunt puţin ridicate, dar în rest totul e bine. Nu ştiu ce trebuie să facem mai departe”, este mesajul transmis părinţilor fetiţei de șase ani îngrijoraţi că nu ştiu ce se întâmplă cu cea mică. Au observat că mezina familiei face pipi în pat pe timpul nopţii şi au decis să facă un control de rutină pentru a se convinge că totul e în regulă.
Transaminazele ridicate pot indica o problemă a ficatului, astfel că au început investigaţiile mai ample în acest sens. „O doctoriţă pediatru ne-a indicat să facem şi creatinkinaza, pentru că ne-a explicat ca la şcoală de unde pot proveni transaminazele ridicate, iar sectorul muşchilor rămăsese neacoperit. Şi la această analiză a ieşit o valoare ridicată. Am fost trimişi la Bucureşti, noi fiind din Constanţa. Am făcut noi teste, de această dată genetice, care au fost trimise la Viena. Fetiţa a fost diagnosticată anul acesta cu boala Pompe. Boala Pompe are la bază o mutaţie genetică. ADN-ul are două copii, una de la mamă şi una de la tată, iar în urma analizelor a ieşit ea are o singură copie cu mutaţie genetică. Noi am început investigaţiile în urmă cu doi ani”, povesteşte Mircea Stanciu, tatăl fetiţiei.
Boala Pompe
Până în acel moment, nici acesta, nici soţia lui nu au auzit vreodată de boala Pompe, astfel că au decis că meargă să discute cu un specialist.
„Am avut şansa să cunoştem la Constanţa o doctoriţă deosebită care acum administrează tratamentul, care ne-a spus să nu mai căutăm specialişti în ţară. Şansa noastră a fost să mergem în Barcelona. Plecarea noastră a coincis cu o vacanţă programată mai demult. Am lăsat vacanţa deoparte. Am ajuns la un spital de copii cu programare pe internet. Am trimis setul de analize. Medicul de acolo şi-a dat seama că este boala Pompe imediat. Ni s-a explicat tot ce trebuia să ştim după această boală. Pentru confirmare ni s-a spus că trebuie să facem un RMN muscular, o analiză a urinei, o biopsie şi un test la efort. Ne-am întors în ţară să facem analizele, dar toţi medicii erau în ceaţă. Nimeni nu ştia nimic, unde se pot face aceste analize. Ne-am întors în Barcelona. Exista degradarea musculară, testul la efort a fost în regulă, dar am zis să facem şi biopsia. Din ce am înţeles noi, pentru a avea acees la tratament, trebuia ca diagnosticul să fie clar. Diagnosticul presupune să ai ambele mutaţii cu defect clare, nu numai una. La Viena au căutat părţile din extremele genomului, dar trebuia căutate şi în cele interioare. Biopsia a venit după trei luni şi diagnosticul a fost pus anul acesta în aprilie. La trei luni după ce am depus dosarul am primit şi tratamentul”, explică tatăl fetei.
Pacienții care au boala Pompe au şansa la o viaţă normală dacă boala se descoperă la timp. „Noi nu am realizat că fetiţa noastră are o problemă. O mai dureau picioarele, dar credeam că s-a jucat mai mult. Nu am crezut că e o problemă extraordinară. Dacă diagnosticul se pune la timp, pacienţii pot avea o viaţă normală, cu urmarea tratamentului”, spune părintele.
Creatinkinaza este o enzimă ce se găseşte în concentraţii crescute în miocard şi muşchii scheletici şi, în concentraţii mult mai mici, la nivelul creierului.
În cazul unui copil care suferă de această boală, unul dintre părinţi trebuie să fie prezent la administrarea tratamentului. Concret, este vorba despre o perfuzie care se face o dată la două săptămâni, o perfuzie care durează cinci ore. Dacă mama este cea care o însoţeşte pe fetiţă la spital de fiecare dată, ceea ce înseamnă automat că trebuie să se învoiască mereu de la serviciu, tatăl a devenit expert în genetică. Urmăreşte toate noutăţile din domeniu, citeşte studii pe această temă şi ştie lucruri pe care până şi specialiştii le ignoră uneori. O astfel de implicare este absolut necesară, pentru că România suferă de o lipsă crasă de specialişti în boli rare. În ţară există puţine centre, iar boala Pompe este o noutate chiar şi pentru acestea. Pentru a putea face ceva pentru bolnavii cu Pompe, părinţii fetei au înfiinţat o asociaţie, care are în prezent opt membri. Unul dintre aceştia, un adolescent în vârstă de 18 ani este primul pacient din România diagnosticat cu boala Pompe.
„Şi în cazul său, la vârsta de doi ani, s-a descoperit că tranzaminatele erau ridicate. Mama copilului a căutat până la vârsta de şase ani un diagnostic în cadrul sistemului nostru de sănătate. Toţi medicii i-au spus că e bine, doar că are tranzaminatele puţin ridicate. Mama în schimb a simţit că ceva nu e bine. Acum 16 ani nu se ştia de boala Pompe, fiind primul caz din România. După diagnosticare a început tratamentul şi acum e bine, doar că e puţin slăbuţ. În cazul acesti boli nu e timp de pierdut!”, explică Mircea Stanciu.
Boala Pompe este o afecţiune neuromusculară rară, progresivă, debilitantă, se transmite autozomal recesiv şi este cauzată de deficitul de acid alfa-glucozidază.
Forma cu debut infantil este caracterizată de progresie rapidă şi speranţă de viaţă foarte scurtă. Decesul survine în general datorită insuficienţei cardiace şi/sau respiratorii înainte de împlinirea primului an de viață. Forma infantilă este rapid progresivă, iar fără tratament duce rapid la deces. Este caracterizată de hipotonie musculară generalizată, areflexie, întârziere în dezvoltarea neuromotorie, macroglosie, hepatomegalie, cardiomegalie hipertrofică, tulburări de alimentaţie şi insuficienţă respiratorie. Forma juvenilă şi adultă se manifestă prin miopatie progresivă cu oboseală, dureri, crampe musculare, fatigabilitate, tulburări de mobilitate cu dificultăţi la mers, afectarea musculaturii faciale cu apariţia unei ptoze palpebrale unilaterale sau bilaterale, dispnee, ortopnee, apnee în timpul somnului şi hipoventilaţie nocturnă. Incidenţa anevrismului cerebral este cu 2,7% mai mare decât în cazul populaţiei generale.
„Părinţii copiilor care suferă de această boală au nevoie de o busolă, de un ghid. Ar trebui ca în setul de analize periodice să fie inclusă şi creatikineaza. Cu toate acestea, speranţa noastră se îndreaptă spre genetică. Problema ar putea fi rezolvată pe această cale”, consideră tatăl fetei.
„Copilul meu de opt ani primeşte bine tratamentul. Problema este că o va face toată viaţa. De curând mi-a spus «Tati, pe mine nu mă deranjează că trebuie să o fac, dar mi-ar plăcea, dacă tot trebuie să o fac toată viaţa să fie o pastilă sau o alifie». Noi sperăm ca medicina să facă progrese în acest sens, iar perfuzia să fie redusă de la cinci ore la două, iar apoi să se transforme într-o pastilă”, spune tatăl fetei.
În cadrul bolilor lizozomale intră mai multe afecţiuni cu transmitere genetică : boala Gaucher, boala Fabry , boala Pompe , boala Nieman-Pick , Mucopolizaharidozele de tip I, II, III si IV. Sunt boli rare, întâlnite la un număr redus de pacienţi, fie din cauza dificultăţii stabilirii diagnosticului, fie pentru că afectează vârste foarte tinere şi din cauza gravităţii şi evoluţiei lor rapide sunt însoţite rapid de decesul pacienţilor.
Boala Gaucher
Oana Monica Dumitrescu suferă de boala Gaucher. Este singura pacientă din România care folosește în prezent un tratament din Israel, întrucât organismul său nu răspunde tratamentului disponibil în România pentru această boală. Șansa ei și a fratelui său a fost un experiment realizat de o echipă de medici din Israel. Este mamă a trei copii sănătoși, dar acest lucru nu ar fi fost posibil dacă se baza doar pe cunoștințele specialiștilor români, care doar i-au pus un diagnostic.
„Am fost diagnosticată la vârsta de doi ani. Atât eu, cât şi fratele meu suntem pacienţi cu boala Gaucher. Până la patru ani, fratelui meu i-au pus tot felul de diagnostice: ciroză, hepatită și altele. Îl tratau pentru simptome. La un moment dat am ajuns la Fundeni şi acolo au văzut că avem boala Gaucher. Ni s-a pus diagnosticul şi atât. Mai mult de atât, acum 34 de ani nu aveau ce să facă. La un moment dat s-a înființat o fundaţie a bolilor lizozomale la Cluj şi ne-am înscris şi noi pentru că deja începusem să ştim şi noi ceva despre boala noastră. În 2004 a venit o echipă de medici din Israel, iar acesta a fost şansa noastră. Dintre toți cei care au fost analizaţi, doar eu, fratele meu şi încă o fată am fost eligibili pentru experimentul respectiv pentru că noi nu aveam splina scoasă, deși și nouă ne-au recomandat mai mulți medici să facem operația respectivă fiind în pericol din cauza mărimii acesteia. Noi am refuzat și acesta a fost până la urmă norocul nostru”, explică Oana. Tânăra care are în prezent 36 de ani este singura din România care face, din două în două săptămâni, tratamentul experimental din Israel, tratament care s-a dovedit a fi un succes în cazul său. Fratele pacientei locuiește în Israel, unde este tratat în continuare.
„Pentru mine este o mare bucurie pentru că sunt un caz experimental reușit. Sunt în formă bună. În exterior arăt bine, dar în interior nu e chiar în regulă. Am fost respinsă la comisia de capacitate a muncii pentru că mă mișc bine și pentru că arăt bine. Analizele mele sunt bune pentru că sunt sub tratament de 14 ani. Mi s-a spus să revin când mă îmbolnăvesc mai rău. Gradul de handicap îl am de 22 de ani, timp în care am fost an de an la comisii, până când au decis într-un final să îmi dea gradul de handicap definitiv”, spune tânăra.
Cea mai mare problemă este faptul că nu există în România un medic specializat în această boală. „În Israel sunt profesori doctori, specializați pe așa ceva. La noi, în România, dacă încep să mă doară oasele, trebuie să merg la ortoped și să îi spun că am boala Gaucher. Acum 14 ani, când nu se cunoștea boala, am făcut un proces inflamator osos foarte puternic, iar medicii mi-au făcut puncție, neînțelegând exact ce am. Mi-au terminat picioarele. Și acum, la unul dinre picioare, glezna este irecuperabilă. Acum așteaptă să se strice și mai tare pentru a-mi pune proteză. Mi s-a spus să trag de timp cât pot, pentru a amâna operația”, povestește pacienta.
Pacienta a fost ajutată foarte mult de cei din Israel. „Avem trei copii, soțul meu a fost testat în Israel. Am avut încredere să avem copii, sub supraveghearea specialiștilor. Copiii sunt probabil doar purtori. Nu au niciun simptom. Am fost foarte ajutată de cei din Israel. La început am făcut un contract pe zece ani și a mers totul strună, dar când acesta s-a terminat și am trecut pe sistemul din România, ne-am împotmolit în birocrație. Am stat șase luni fără tratament, iar starea mea s-a înrăutățit. Cei din Israel mi-au dat până la urmă tratamentul prin donație. La un moment dat am început să solicit să fie acordat și în România tratamentul, pe baza unui protocol. S-a aprobat în sfârșit și acest protocol. Mai sunt piedici, dar încercăm să le rezolvăm încet, încet. Cu toate acestea, nu există medici specialiști în țară. Am simțit la un moment dat nevoia să îmi îmunătățesc starea de sănătate și am vrut să apelez la ceva suplimente. Tratamentul este bun, dar trebuie susținut sistemul osos. Procesul de evoluție a bolii nu s-a oprit. Orice supliment aș lua, trebuie să fie acordat ținându-se cont de boala mea și aici ne împotmolim din nou”, spune Oana.
Împreună cu soțul său, Oana a dezvoltat o afacere proprie, soluție care îi permite să muncească în propriul ritm, ceea ce ca un simplu angajat nu ar fi posibil, din cauza durerilor osoase incontrolabile și a oboselii care intervine când se așteaptă mai puțin. „Cine ne angajează pe noi, dacă la amiază simțim nevoia sa ne întindem sau când trebuie ca o dată la două săptămâni să pierdem o zi pentru a merge la perfuzii? Eu într-adevăr am avut un angajator care m-a înțeles, care a fost empatic cu problema mea. Cu toate acestea, am ales să avem propria afacere pentru a nu depinde de bunăvoința nimănui”, explică Oana.

Pentru un pacient care suferă de o boală rară, boala nu mai este rară, ci este realitatea cu care se confruntă în fiecare zi.
Boala Gaucher apare când anumite lipide se acumulează în mod excesiv la nivelul ficatului, splinei, măduvei osoase și, mai rar, a creierului. Aceasta acumulare de țesut adipos interferă cu funcționarea normală a organelor și poate determina mărirea în volum a organului și dureri osoase.
Boala Gaucher apare datorită unei deficiențe enzimatice și uneori termenul de “deficiența de glucocerebrozidază” este folosit pentru a descrie afecțiunea. Poate apărea la orice vârstă și afectează femeile și bărbații în proporții egale.
Semnele și simptomele bolii Gaucher variază larg de la un pacient la altul. Durerile osoase sau fracturile sunt deseori primul simptom. Simptomele pot fi: anormalități scheletale, incluzând subțierea unor oase, dureri osoase și fracturi, mărirea în volum a ficatului sau a splinei sau a ambelor, anemie, datorată prezenței unei cantități prea mici de hematii sănătoase, o sensibilitate mare la vânătăi, deteriorare cognitivă, incluzând retardare mentală sau demență, pete galbene la nivelul ochiului, mișcări oculare anormale, funcționare anormală a rinicihilor și plămânilor, colorarea maronie a pielii.
Dacă medicul suspectează prezența bolii Gaucher sau dacă aceasta este prezentă la membrii familiei, diagnosticul se pune pe teste de sânge care evaluează nivelul enzimei asociate cu aceasta boală.
[stextbox id=’custom’]
Ce înseamnă o boală rară?
O boală sau o tulburare este definită ca fiind rară în Europa, atunci când afectează mai puțin de o persoană din 2000. O boală sau o tulburare este definită rară în SUA, atunci când aceasta afectează mai puțin de 200.000 de americani, la un moment dat.
80% din bolile rare au cauză genetică. 50% din aceste boli afectează copiii. Peste 95% din bolile rare nu au încă tratament nicăieri în lume.
În România trăiesc peste 1.000.000 de pacienţi afectaţi de boli rare. Potrivit datelor Alianței Naționale pentru Boli Rare, în România 75% dintre cei care suferă de o boală rară sunt copii, iar 9 din 10 pacienți nu beneficiază de un tratament adecvat pe fondul lipsei diagnosticului sau a diagnosticării tardive. Situația este și mai complicată dacă se ia in considerare faptul că bolile rare au un impact profund și asupra familiilor pacienților. Majoritatea sunt încă nediagnosticați. Alţii sunt diagnosticați greșit sau incomplet; sunt izolaţi și vulnerabili. Au nevoie de o abordare specifică și multidisciplinară pentru a le asigura serviciile de care au nevoie, pentru a crește calitatea vieţii lor. Serviciile sunt insuficiente la nivel național, iar problematica abordării bolilor rare este complexă și interdisciplinară. Faptul că pacienții cu boli rare sunt răspândiți la nivel național, în diferite regiuni, necesită o intervenție centralizată. Lipsa politicilor specifice de sănătate privind bolile rare și deficitul de expertiză în domeniu au drept consecință diagnosticarea întârziată și accesul dificil la asistența medicală. Această situație are drept rezultate neajunsuri suplimentare de natură fizică, psihologică și intelectuală, tratamente inadecvate sau chiar dăunătoare.
Conform EURRDIS (Organizația Europenă de Boli Rare) în Europa există peste 8.000 de boli rare care afectează peste 30 de milioane de pacienți în U.E.
Dezvoltarea de medicamente poate reprezenta o provocare în cazul bolilor rare. Datorită numărului mic de pacienți, companiile farmaceutice întâmpină greutăți la înrolarea numărului potrivit de pacienți pentru un studiu clinic riguros. În plus, cunoștințele științifice despre bolile rare și experința cu acestea sunt limitate. Din aceste motive, pentru majoritatea bolnavilor nu există tratamente aprobate.
Bolile rare afectează, în România, peste 1 million de persoane. Aceste persoane sunt, în marea lor majoritate, marcate de excluziune socială și sărăcie, trăind în parametri de viață precari, cu acces redus la servicii de diagnosticare precoce, terapii și tratamente cu medicamente orfane, teste genetice, screening și servicii sociale de suport.
[/stextbox]
 RO_Landscape - Felicitare Craciun - banner.png)

 OPINIE | Să iubim Crăciunul, pentru că e miracolul vieţii
OPINIE | Să iubim Crăciunul, pentru că e miracolul vieţii Peste câteva ore doar, vom simți cu toții bucuria Naşterii Lui. Și vine vremea
 De la un palaneț împărțit la trei, în România, la farfuria cu escargots la Ritz
De la un palaneț împărțit la trei, în România, la farfuria cu escargots la Ritz Cineva mi-a reprodus discuția dintre trei tineri studenți care coborau de la
 Liberalii oficializează segregarea şcolară, în siajul gândirii USR
Liberalii oficializează segregarea şcolară, în siajul gândirii USR Guvernul a identificat două nevoi reale. Pe de o parte, copiii au nevoie să fie
 Congresul PNL. Cine strică marfa o plăteşte
Congresul PNL. Cine strică marfa o plăteşte Nu daţi vrabia din mână, oameni buni, pe cioara nebună de pe gard, îi conjură
 Apocalipsa după UNTOLD. Pista de atletism distrusă, iar gazonul de un milion de euro e înlocuit cu unul de 180.000
Apocalipsa după UNTOLD. Pista de atletism distrusă, iar gazonul de un milion de euro e înlocuit cu unul de 180.000 An după an daunele materiale provocate de cel mai mare festival de muzică


 Profesii care vă permit să călătoriți și să munciți: oportunități și perspective
Profesii care vă permit să călătoriți și să munciți: oportunități și perspective Experții de la agregatorul de joburi Jooble ne-au descris 10 profesii de perspectivă, care vă
 Top 5 motive să achiziționezi o Skoda Karoq
Top 5 motive să achiziționezi o Skoda Karoq Într-o piață plină de opțiuni atrăgătoare, Skoda Karoq se evidențiază prin oferirea
 Trenduri inovatoare în designul meniurilor pentru restaurant. Ce este de interes în industria ospitalității?
Trenduri inovatoare în designul meniurilor pentru restaurant. Ce este de interes în industria ospitalității? Atunci când sunteți în căutarea unei mape de meniu potrivite, care să dureze, fiecare
 Clujul s-a asociat cu județele Alba și Bihor pentru dezvoltarea turistică a Țării de Piatră din Apuseni
Clujul s-a asociat cu județele Alba și Bihor pentru dezvoltarea turistică a Țării de Piatră din Apuseni Consilierii județeni au adoptat astăzi proiectul de asociere pe 10 ani între județele Alba,
 Ofertă irezistibilă pentru un sejur la mare. A început ”Litoralul pentru toți”
Ofertă irezistibilă pentru un sejur la mare. A început ”Litoralul pentru toți” Românii pot petrece, începând de astăzi, un sejur pe litoralul românesc cu preţuri ce
 Arteră spre inima Balcanilor (III): Bosnia și Herțegovina, încremenită în proiect
Arteră spre inima Balcanilor (III): Bosnia și Herțegovina, încremenită în proiect 

Nepoțelul meu a fost diagnosticat cu Sindrom Marshall. Ma interesează tot ce tine de aceasta boala,tratament si rezultate.
G . C. 30 ani am fost diagnosticat cu boala pompe tardiva de anul trecut …am un an de cand primesc tratamientul in Germanía …
In Romania tratamientul este oferit pt cei care sufera de aceasta boala rara?